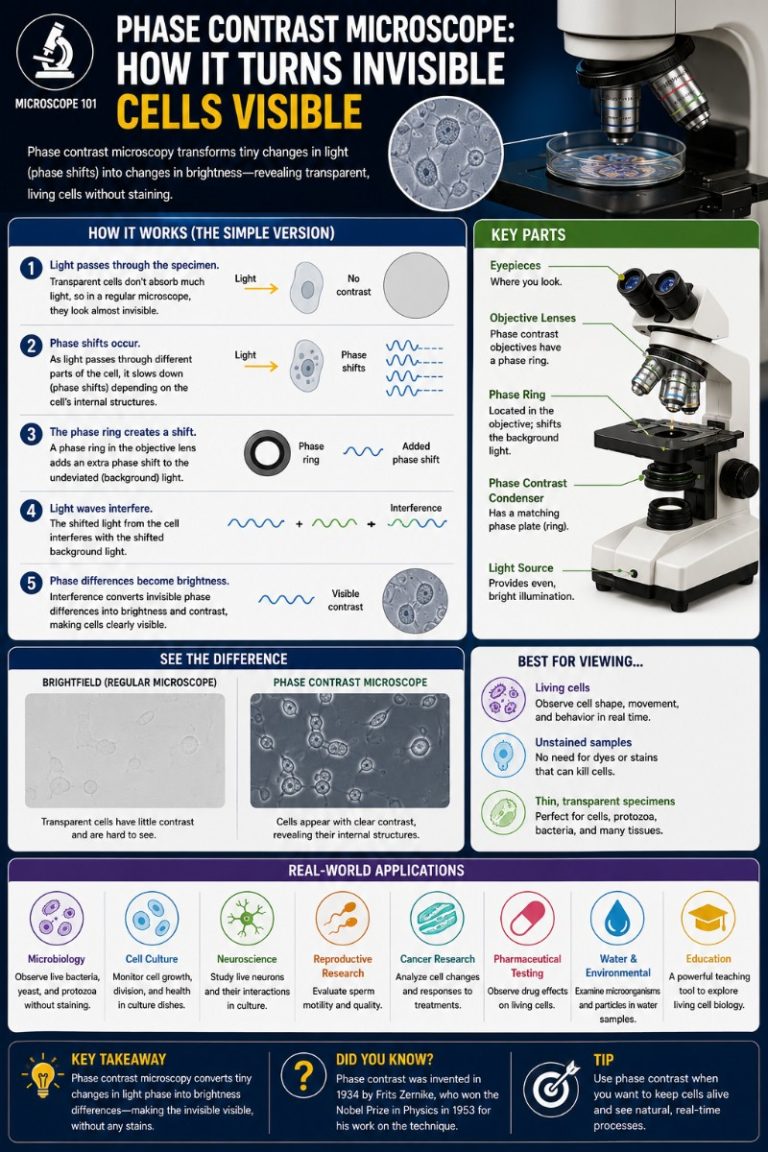
A phase contrast microscope makes living, unstained, transparent specimens visible by converting invisible differences in light’s phase into visible differences in brightness — something a standard bright-field microscope simply cannot do. It achieves this through two matched optical components: an annular diaphragm in the condenser and a phase plate in the objective, which together exploit destructive interference to darken dense cell structures against a lighter background. The result is crisp, high-contrast images of living cells without staining, fixation, or killing the specimen.
How a Phase Contrast Microscope Works (Step by Step)
The magic of phase contrast lies in understanding what light does — and doesn’t do — when it passes through a transparent specimen. Here is the complete light path, from condenser to eye.
The Problem It Solves: Phase vs. Amplitude
Your eyes and a camera sensor both detect two properties of light: amplitude (brightness) and wavelength (color). What they cannot detect is phase — the timing or position of the light wave within its cycle.
Most living biological specimens are phase objects. They are nearly transparent — they transmit almost all incoming light without absorbing it, so amplitude barely changes. Under a standard bright-field microscope, a living cheek cell or bacterium looks like a nearly empty, washed-out smear. There is real structure there — but no contrast to make it visible. You would normally have to stain the specimen, a process that often kills living cells and can distort their natural shape.
Phase contrast sidesteps that problem entirely. Transparent structures do shift light’s phase — they just don’t change its brightness. The phase contrast microscope catches that invisible phase shift and converts it into a brightness difference the eye can see.
Optical Path Length and the Quarter-Wavelength Phase Shift
The physics hinge on a simple relationship: Optical Path Length (OPL) = refractive index (n) × physical thickness (d). Light travels more slowly through materials with a higher refractive index.
Cell structures — nuclei, organelles, membranes — have a higher refractive index than the surrounding water or buffer (cell cytoplasm n ≈ 1.35–1.38 vs. water n ≈ 1.33). Light passing through a cell nucleus is retarded: it arrives at the far side of the cell slightly behind the light that passed through the surrounding medium. For a typical mammalian cell, this retardation amounts to roughly one-quarter of a wavelength (λ/4, about 90°). That tiny timing difference is invisible on its own — it doesn’t change how bright the wave looks. It’s a phase shift with no visible consequence — yet.

The Annular Diaphragm and Phase Plate: Converting Phase to Contrast
This is where the two key components of phase contrast come in. They work as a matched pair — in fact, they must be matched to the same Ph number (Ph1, Ph2, or Ph3) to function at all.
Step 1 — The annular (ring) diaphragm. Located in the condenser, this ring-shaped aperture replaces the standard circular aperture. Instead of flooding the specimen with a filled cone of light, it produces a hollow cone — only a ring of light passes upward through the specimen.
Step 2 — Light splits at the specimen. When this hollow cone of light hits the specimen, it splits into two components:

- Undeviated (direct) light: passes straight through without being scattered. It carries no information about the specimen’s structure. It arrives at the objective’s back focal plane in the same ring shape as it left the condenser annulus.
- Diffracted light: scattered by structures in the specimen. This light spreads out across the full back aperture of the objective — and it carries the ~λ/4 phase retardation from the specimen’s refractive index differences.
Step 3 — The phase plate in the objective. At the back focal plane of the objective lens, a transparent glass plate has a ring-shaped zone etched or coated into it. This phase ring is positioned precisely to coincide with the image of the condenser annulus — meaning the undeviated direct light passes through it, while the diffracted light largely misses it and passes through the surrounding clear glass.
The phase ring does two things to the direct light:
- It advances or retards it by another λ/4 (90°).
- It attenuates it — a gray absorbing coating dims the bright direct light so its intensity better matches the much fainter diffracted light. Without this dimming step, the direct light would simply overwhelm the diffracted signal.
Step 4 — Destructive interference. The direct light has now been shifted by λ/4 by the phase plate. The diffracted light was already shifted ~λ/4 by the specimen. When both waves arrive at the image plane and recombine, they are approximately ½ wavelength (180°) out of phase — and they destructively interfere. The result: wherever a denser specimen structure was, the combined wave has reduced amplitude — it appears darker than the surrounding background. Phase differences have been converted into brightness differences the human eye can detect.
This standard configuration is called positive (dark) phase contrast: denser structures appear dark on a lighter gray background. A less common variant, negative (bright) phase contrast, reverses this so structures appear bright.
Key Definitions: Phase Shift, Optical Path Difference, and Contrast
Phase contrast microscopy has a specific vocabulary. Here are the core terms defined precisely so they’re unambiguous:
- Phase: The position of a wave within its oscillation cycle, measured in degrees (0°–360°) or fractions of a wavelength. A phase difference of 180° (½λ) means the peaks of one wave align with the troughs of another.
- Optical path length (OPL): The effective distance light travels, accounting for the medium it passes through: OPL = n × d (refractive index × physical thickness). Two rays from the same source that pass through different materials can have different OPLs — and therefore arrive with a phase difference.
- Optical path difference (OPD): The difference in OPL between two rays — in phase contrast, between the direct ray and the diffracted ray. It’s the OPD that becomes the phase shift.
- Contrast: The difference in brightness between the specimen and its background. Phase contrast converts an invisible OPD into visible contrast by making constructive and destructive interference produce brightness variation.
- Direct (undeviated) light: The portion of the illuminating beam that passes through the specimen unscattered. Carries no specimen information.
- Diffracted light: The portion scattered by structures in the specimen. Carries specimen information, including the phase shift caused by refractive index differences.
- Positive vs. negative phase contrast: In positive (the default), dense structures appear darker than the background. In negative, they appear brighter. The phase plate is designed differently for each.
Why Phase Contrast Images Have Halos
If you’ve used a phase contrast scope and noticed a bright glowing outline — almost like a fluorescent rim — wrapping every cell and particle, you’ve met the most famous artifact in phase contrast microscopy: the halo.
The halo is not a mistake. It’s an inherent optical artifact caused by the imperfection of the phase plate separation. The annular diaphragm and phase ring can’t achieve a perfect split: some diffracted light inevitably passes through the phase ring (where it gets attenuated and phase-shifted along with the direct light), and some direct light spills outside the phase ring. This means the phase manipulation at sharp specimen edges is incomplete, and the recombination of light at boundaries produces a bright fringe — the halo — surrounding every edge.
In practice, this matters because halos can obscure fine edge detail. Beginners sometimes interpret the glowing rim as a real biological structure — a “thick membrane” or “bright coating” — when it’s actually the artifact. Once you know to look for it, it becomes diagnostic: anything with a bright glowing border is phase contrast doing its job, not a feature of the cell.
A related artifact is shade-off: in large, uniform objects, the center appears closer to background brightness than the edges, so contrast “shades off” toward the middle. This is why phase contrast is better suited to thin, small structures than large, thick, homogeneous regions.
Reducing halos: Apodized phase contrast objectives (available from Nikon and others) incorporate a neutral-density absorber at the outer edge of the phase ring that grades the transition between the phase ring and the open aperture. This dramatically reduces halo intensity while preserving contrast in the specimen itself.
Phase Contrast vs. Bright-Field vs. DIC
Choosing the right contrast method depends on your specimen, your hardware budget, and what you can tolerate in terms of artifacts. Here’s how the three main methods compare:
| Feature | Bright-Field | Phase Contrast | DIC |
|---|---|---|---|
| Contrast on live, unstained cells | Poor | Excellent | Excellent |
| Staining required? | Usually yes | No | No |
| Halo artifact | None | Yes (inherent) | None |
| Pseudo-3D appearance | No | No | Yes (“shadow-cast”) |
| Works with plastic culture dishes | Yes | Yes | No (birefringence) |
| Thick specimens | Moderate | Poor (shade-off) | Better |
| Cost / complexity | Low | Moderate | High (polarizers + prisms) |
When to pick phase contrast over DIC: The single biggest practical reason labs default to phase contrast for cell culture is plastic. Standard polystyrene culture dishes and flasks are birefringent — they interfere with the polarized light that DIC requires, making DIC essentially useless through them. Phase contrast works fine through plastic, which is why virtually every inverted microscope in a tissue culture room uses phase contrast as its primary imaging mode. If you’re watching cells grow in a T-flask or a multi-well plate, phase contrast is your tool.
DIC is preferable when you need a genuine 3D surface rendering, when halos are unacceptable for precise edge measurement, or when imaging thicker biological specimens on glass slides. For a deeper look at how illumination methods compare, see our guide to dark field microscopy as another contrast-enhancing alternative.
Bright-field remains the right choice when your specimen is stained, fixed, or naturally absorbs light — it’s simpler and cheaper, with no special hardware beyond a standard compound light microscope.
What Phase Contrast Microscopy Is Used For
The core use case is straightforward: any transparent, unstained, living specimen that you can’t or don’t want to stain. In practice, that covers an enormous range of work:
- Live cell imaging and cell culture monitoring: Watching cells divide, migrate, or die in real time. Phase contrast is the standard for monitoring cell health in incubators — checking confluence, spotting contamination, counting mitotic figures — without disturbing the culture. Olympus Life Science offers a detailed technical primer on the technique for those who want to go deeper.
- Bacteria: Most bacteria are too small and transparent for comfortable bright-field viewing without staining. Phase contrast reveals bacteria under a microscope as crisp dark rods, cocci, or spirals against a light background, and shows motility clearly.
- Protozoa and pond water: Amoeba, paramecia, and other protists in pond water show striking internal detail — pseudopods, food vacuoles, and cilia — without any preparation beyond a wet mount.
- Sperm motility analysis: Clinical andrology labs use phase contrast to assess sperm motility and morphology on live, unfixed samples. The dark sperm heads on a pale background are easy to track and count.
- Unstained tissue sections: Thin tissue sections can be examined for structure without the time cost of staining protocols, useful during intraoperative pathology or when screening before selecting sections to stain.
- Mycology: Fungal hyphae and spores in wet mounts show excellent contrast and internal organization under phase contrast.
The common thread: specimens that are alive, thin, and optically transparent. If your sample fits that description, phase contrast should be your first consideration before reaching for stains. For a broader comparison of imaging approaches for different sample types, our overview of types of microscopes covers when each platform applies.
What You Actually See: First-Hand Notes from the Eyepiece
Reading about phase contrast is one thing; knowing what to expect when you actually sit down at the scope is another. Here’s what the experience looks like in practice.
Switch from bright-field to phase contrast on a slide of living cheek cells and the transformation is immediate: what was a nearly empty, washed-out gray field suddenly fills with structure — cell outlines, nuclei, and cytoplasmic granules “pop” into view in shades of gray, like an image developing in a darkroom. In positive phase contrast, cells appear as soft gray discs with a darker, denser nucleus surrounded by granular cytoplasm. Dense structures are darker; the background is a lighter, even gray.
Every edge you see will wear the characteristic bright halo — a thin glowing rim. It’s pretty, but it will smear the sharpest edge details. Beginners almost universally misidentify it as a thick cell membrane on first viewing. It isn’t. Once you know what halos look like, you stop being fooled.
Moving specimens — swimming bacteria, protozoa, sperm — appear as crisp dark bodies darting across a pale field. Motility is dramatically easier to see than in bright-field, where they can disappear entirely against the background.
Three beginner mistakes and how to fix them:
- Forgetting to align the rings. If you get low contrast or a washed-out image, the condenser annulus probably isn’t centered on the objective’s phase ring. Pull out the centering telescope (or use a Bertrand lens) and overlap the bright condenser ring onto the dark phase ring. This is the single most common setup error.
- Mismatched Ph numbers. Phase contrast objectives are marked Ph1, Ph2, or Ph3 — the number specifies which size condenser annulus they’re designed to work with. Running a 40× Ph2 objective with the condenser turret set to Ph1 gives weak, incorrect contrast. Always match the condenser turret to the objective’s Ph marking.
- Leaving the condenser annulus in during bright-field. If the condenser turret is on a Ph position when you switch to a non-phase objective, the image goes dark and patchy. Turn the turret to the “BF” (open) position for standard bright-field work.
Beyond setup errors, thick specimens cause trouble: phase contrast works best on thin, near-transparent samples. A dense clump of pond debris or a thick tissue section goes muddy and flat because the technique assumes a small, well-defined phase shift — not the complex overlapping shifts from thick material. If your specimen is thick, DIC or fluorescence will serve you better. Also make sure to set up Köhler illumination first, then align the phase rings — a dirty or poorly-focused condenser kills phase contrast contrast just as surely as a misaligned annulus.
Frequently Asked Questions
Who invented the phase contrast microscope?
Dutch physicist Frits Zernike invented the phase contrast microscope in the 1930s. He had worked out the theory of phase shifting in the late 1920s and built the first working instrument around 1932. He was awarded the Nobel Prize in Physics in 1953 specifically for this invention, which the Nobel Committee described as having “great practical importance in biology and medicine.” Zeiss began commercial production of phase contrast microscopes in the 1940s.
What exactly is a phase plate, and where is it located?
A phase plate is a glass disc with a ring-shaped zone — the phase ring — that is either etched to a slightly different thickness or coated with a thin dielectric or metallic film. It is mounted at the back focal plane of the objective lens, not in the condenser or eyepiece. The phase ring is precisely sized to coincide with the focused image of the condenser annulus so that only the undeviated direct light passes through it. The ring advances or retards that direct light by λ/4 and attenuates it, setting up the destructive interference that creates contrast.
Do you need to stain specimens for phase contrast?
No — and avoiding staining is the whole point. Phase contrast is specifically designed for unstained, living specimens. It creates contrast from the specimen’s own optical properties (refractive index differences) without adding any dye or fixative. This means cells remain alive and behaving naturally throughout imaging. You can stain a specimen and then use phase contrast, but it’s unnecessary and the stain’s added absorption can interfere with the pure phase mechanism.
Can a regular microscope be converted to phase contrast?
Not with a filter or a simple attachment. Phase contrast requires two matched hardware upgrades: special phase contrast objectives (marked Ph1, Ph2, or Ph3) and a matching annular diaphragm condenser (or a phase contrast condenser turret). Some manufacturers sell retrofit phase contrast condenser kits for existing microscopes, provided the objective mount and condenser mount are compatible. If your scope’s objectives are standard bright-field lenses, no optical accessory will add phase contrast — you need the phase ring physically built into the objective. Check our guide to parts of a compound microscope to understand which components would need replacing.
How much does a phase contrast microscope cost?
Entry-level research-grade phase contrast microscopes (upright, for fixed specimens and wet mounts) start around $500–$1,500 for basic educational models. Mid-range lab-quality upright phase scopes run $2,000–$6,000. Inverted phase contrast microscopes for cell culture monitoring — the standard in biology labs — typically cost $3,000–$15,000+ depending on the number of phase objectives and imaging accessories. High-end research systems (motorized, with camera, software, and apodized objectives) can exceed $30,000. Used and refurbished units from Nikon, Olympus, Leica, and Zeiss offer significant savings.
Why does phase contrast work poorly on thick specimens?
Phase contrast assumes that the phase shift from the specimen is small and relatively uniform — around λ/4 from a thin, transparent layer. In a thick specimen, light passes through many layers of varying refractive index and picks up large, complex, overlapping phase shifts. The phase plate can only handle a specific, narrow range of phase shift; large or uneven shifts fall outside that range and produce muddy, low-contrast images with severe shade-off. Thick specimens are better handled by DIC, confocal fluorescence, or by sectioning before imaging.
Does phase contrast affect magnification or resolution?
No. Phase contrast affects contrast only — not magnification and not optical resolution. A common misconception is that it “improves the image” in the sense of making it sharper; what it actually does is make structures visible that were already there but invisible due to lack of contrast. True resolution is governed by numerical aperture and wavelength, and the phase plate can actually slightly reduce effective NA by blocking some of the aperture. See our explainer on magnification vs. resolution in microscopy for the full distinction.
Conclusion
Phase contrast microscopy solves a fundamental optical problem: transparent specimens shift light’s phase but not its brightness, making them invisible to the naked eye and to standard bright-field optics. By pairing an annular diaphragm with a matched phase plate, the microscope intercepts the direct and diffracted light separately, applies a precise λ/4 shift to the direct beam, and then lets destructive interference do the work — turning a hidden phase difference into a visible brightness difference. The result is real-time, artifact-rich, stain-free imaging of living biology that has remained indispensable in labs for nearly a century.
Have you tried phase contrast on your own scope — or noticed the halo for the first time and wondered what it was? Drop a question or share what you were looking at in the comments below. We’d love to hear what specimens have surprised you most.