
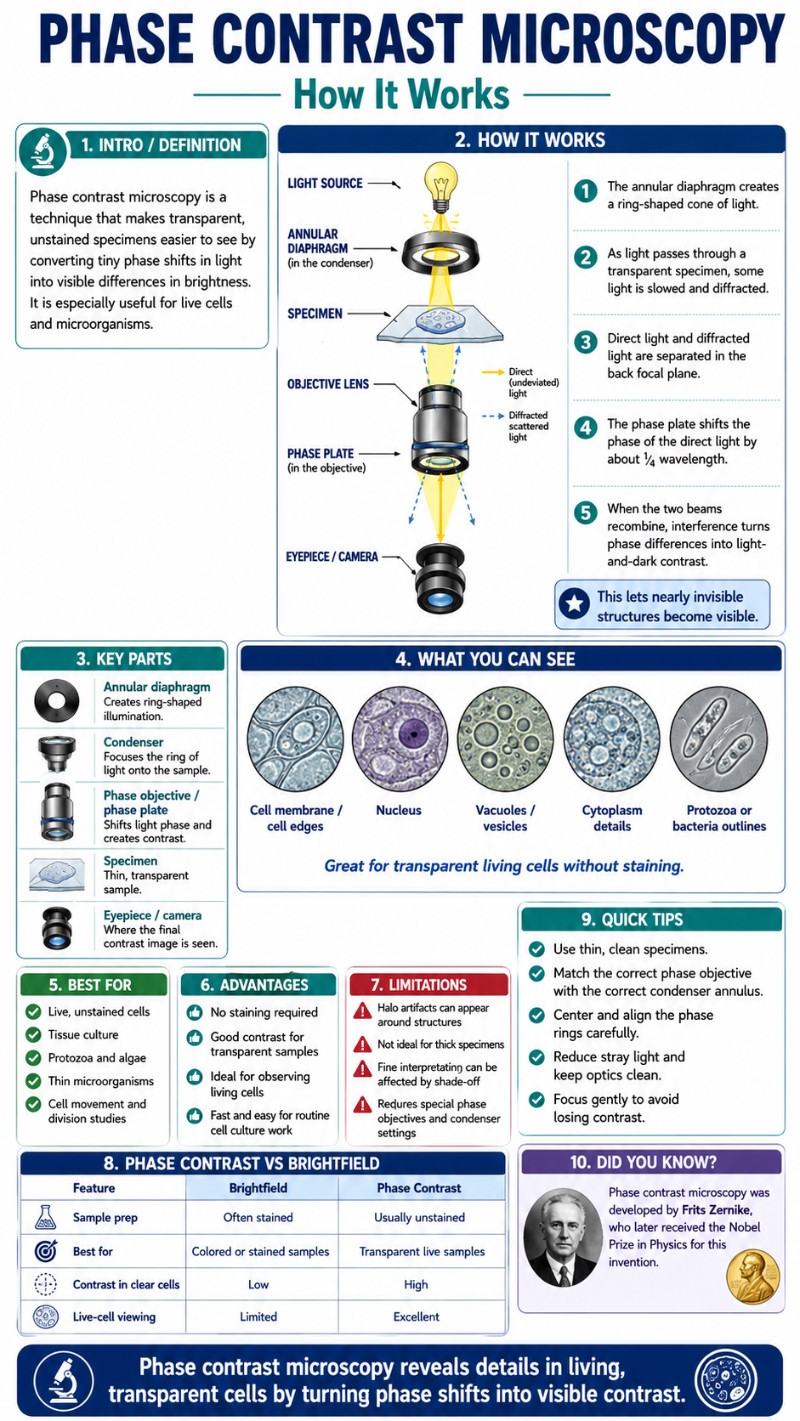
A phase contrast microscope makes transparent, unstained, living cells visible by converting invisible differences in refractive index — called phase shifts — into visible differences in brightness. It does this using two matched optical components: an annular diaphragm in the condenser and a phase plate inside the objective. The result is crisp, gray-on-light contrast of live specimens that brightfield illumination leaves nearly invisible, with no staining, no fixation, and no dead cells.
The Problem Phase Contrast Solves — Why Living Cells Are Nearly Invisible
To understand why the phase contrast microscope matters, you first have to appreciate why a brightfield microscope fails so badly on living material. Brightfield creates contrast the only way it can: by detecting differences in how much light a specimen absorbs. A cell that absorbs a lot of light looks dark; one that absorbs little looks nearly the same as the background — pale and washed out.
Living, unstained cells absorb almost no light. They are what optical physicists call phase objects — transparent structures that don’t block light, but do alter its speed. Light slows down slightly when it travels through denser material, and the interior of a cell (refractive index roughly 1.36–1.40) is denser than the surrounding water or culture medium (refractive index ~1.33). So the light rays that pass through the cell emerge a fraction of a wavelength behind those that passed through the clear medium on either side. That delay is a phase shift. The human eye and a camera are completely blind to phase differences — they only respond to amplitude (brightness) and color. This is why, under standard brightfield, you can squint at a culture dish and barely make out a faint ghost outline of cells that are sitting right in front of you.
The solution Frits Zernike devised in the early 1930s was to trick the optics into converting that invisible phase difference into an amplitude difference the eye can see.
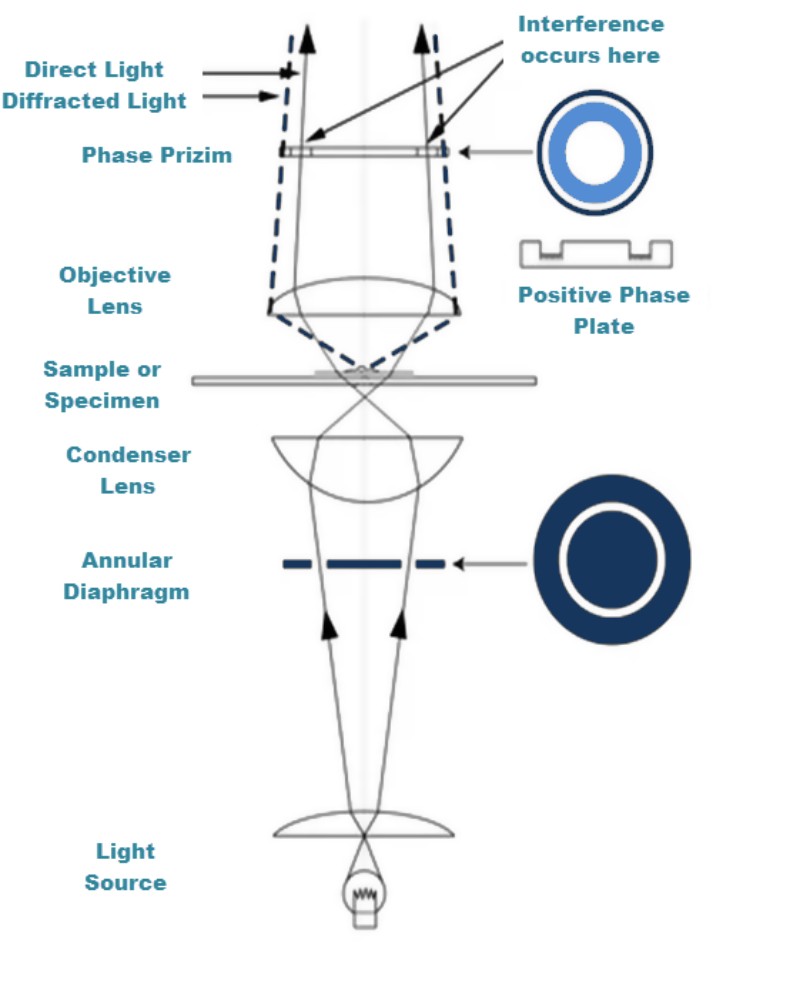
How Phase Contrast Works, Step by Step
The mechanism runs in four stages, each building on the last.
Direct Light vs. Diffracted Light
When light enters a specimen on the stage, it splits into two populations. Most of it passes straight through without being bent — this is called direct or surround light (the S-wave). A smaller fraction is scattered and bent by the structural detail inside the specimen — this is diffracted light (the D-wave). In an empty field of view with no specimen, almost all the light reaching the eyepiece is direct light.
The Quarter-Wavelength Shift and Zernike’s Trick
Here is the insight that won Zernike the 1953 Nobel Prize in Physics: diffracted light, simply by having interacted with the specimen, is naturally retarded by about ¼ wavelength (90°) relative to the direct light. Two waves that are ¼ wavelength apart don’t produce destructive interference — they don’t cancel each other. But if you could shift the direct light by another ¼ wavelength, the direct and diffracted waves would be ½ wavelength (180°) out of phase. At 180°, they destructively interfere: where the two waves recombine, they partially cancel, and the specimen appears darker than the background. An invisible phase difference has become a visible brightness difference.

The phase plate inside the objective does exactly that — it introduces an additional ¼λ shift to the direct light, bringing the total offset between direct and diffracted light to ~½λ and triggering destructive interference wherever specimen detail exists.
Why the Direct Light Is Also Dimmed
There’s a second problem. Direct light vastly outnumbers diffracted light in amplitude. If you just shift the direct light and let both waves recombine at their natural intensities, the interference effect is swamped and the contrast remains poor. The phase ring in the objective is therefore also partially absorbing — it’s gray-tinted — which dims the direct beam down so its amplitude better matches the weaker diffracted beam. Only when the two waves are closer in amplitude does their interference produce the maximum visible contrast difference.


The Two Key Components — Annular Diaphragm and Phase Plate
Everything in the mechanism above depends on two matched parts working in concert. Understanding each one makes setup and troubleshooting much clearer.
The annular diaphragm (also called the phase annulus or annular stop) sits in the front focal plane of the microscope’s condenser — not where the normal iris diaphragm sits, but in a rotating turret below it. Instead of a round open aperture, it is a ring-shaped slit. Light passes only through that ring, producing a hollow cone of illumination directed up through the specimen. This matters because it separates the paths of direct and diffracted light in a way that lets the phase plate act on the direct beam selectively.
The phase plate sits in the rear focal plane of the objective — inside the objective itself. It contains a ring of glass or coating whose diameter matches the image of the condenser annulus at that plane. The ring does two things: (a) it shifts the direct beam by ¼λ via an optical-path-difference coating, and (b) it attenuates the direct beam through partial absorption. Diffracted light, scattered at wider angles by the specimen, misses the ring and passes through the clear glass of the objective unaltered.
Critical alignment: The image of the condenser annulus must be precisely superimposed on the phase ring in the objective. This is why phase contrast objectives are engraved with markings like Ph1, Ph2, or Ph3 — each number corresponds to a different annulus size on the condenser turret. A Ph1 objective must be paired with the Ph1 annulus position; a Ph2 with Ph2, and so on. A mismatch means the two rings don’t overlap, and the phase effect collapses. For a broader overview of how microscope objective lenses are classified, see our dedicated guide.
Setting Up and Aligning Phase Contrast
Getting phase contrast right takes a few minutes the first time, and almost no time once you know the steps.
- Set up Köhler illumination first. Phase alignment depends on a focused condenser; a poorly set condenser height ruins the ring overlap before you even start.
- Select the correct objective and rotate the condenser turret to the matching annulus position (Ph1/Ph2/Ph3 as marked).
- Center the rings. Remove an eyepiece and insert a centering telescope (also called a Bertrand lens or phase telescope). You’ll see a bright ring (the illuminated annulus) and a darker gray ring (the phase ring). Use the condenser annulus centering screws to slide the bright ring until it sits exactly over the darker one. When the two rings are concentric, the phase effect is at maximum contrast.
- Re-insert the eyepiece and focus on your specimen.
The most common beginner mistake is switching to a phase objective and assuming the condenser is already set up correctly — leaving it on the open-field brightfield position. The image looks like dim, muddy brightfield. Fix: rotate the condenser turret to the correct Ph setting. The second most common mistake is skipping the centering step; uncentered rings produce washed-out, uneven contrast that looks like it isn’t working at all.
Positive vs. Negative Phase Contrast
There are two flavors. In positive phase contrast — by far the most common type in biology labs — the phase ring advances the direct light by ¼λ, so dense cell structures appear darker than the gray background. The nucleus shows up as a dark oval, the cytoplasm as a lighter gray, and the cell membrane as a defined dark rim. This is what most people picture when they say “phase contrast.”
In negative phase contrast, the direct light is retarded instead of advanced, flipping the interference result: dense structures appear brighter than the background. Negative phase contrast is less common but can be useful for certain specimens where the dark-on-light rendering of positive phase creates confusion with overlapping structures.
What You Can See — Best Specimens and Applications
The transformation you see when you switch from brightfield to phase contrast on a live slide is startling. On a cheek-cell preparation, cells that were barely a pale outline suddenly “pop” into crisp gray shapes with a visible nucleus, defined membrane, and hints of cytoplasmic granules. The background goes from bright white to an even, flat gray. And around every cell edge you’ll notice a thin bright glow — the characteristic halo — which is normal and discussed below.
Viewing bacteria is one of phase contrast’s best-known applications: small dark rods or cocci visible against the gray field, many of them actively motile — tumbling and darting in real time — with no stain and no killed specimen. Live protozoa in pond water show even more dramatic detail: the nucleus, contractile vacuole, and food vacuoles of an amoeba or paramecium are all visible, and you can watch digestion and locomotion in real time.
Phase contrast is the workhorse for:
- Live cell culture monitoring (mitosis, cell spreading, morphology changes)
- Bacterial motility and morphology studies
- Protozoa, algae, and other microorganisms in aqueous media
- Unstained thin tissue sections where you need to identify structure before committing to a stain
- Sperm motility analysis in reproductive biology
Phase contrast works best on thin specimens in aqueous media. Very thick specimens or high-refractive-index mounting media exaggerate halos and cause large structures to look washed out in the center — a phenomenon called “shade-off.” For those cases, differential interference contrast (DIC) is often a better choice, though at higher cost.
Phase Contrast vs. Brightfield vs. DIC
Here’s how the three main contrast modes stack up for practical use:
| Feature | Brightfield | Phase Contrast | DIC (Nomarski) |
|---|---|---|---|
| Specimen prep needed? | Usually stained | None — live specimens | None — live specimens |
| Source of contrast | Light absorption | Phase shifts → destructive interference | Optical path gradient → pseudo-3D relief |
| Best for | Stained histology, colored specimens | Live cells, bacteria, culture work | Live cells, surface detail, thick specimens |
| Halo artifact? | No | Yes — inherent | No |
| Relative cost | Lowest | Moderate | Highest |
| Key limitation | Kills or obscures living specimens | Halo artifact; shade-off in thick areas | Cost; birefringent specimens distort |
Phase contrast occupies the sweet spot for most teaching labs and research setups: it reveals live, unstained specimens with genuine structural detail, costs far less than DIC, and requires no polarizers or Wollaston prisms. DIC produces a beautiful halo-free pseudo-3D image but adds significant cost and complexity. Darkfield microscopy is sometimes confused with phase contrast, but it works on a completely different principle — oblique illumination that makes specimens glow bright against a black field — and reveals surface features rather than internal structure.
Limitations and the Halo Effect
Phase contrast is not a perfect solution for every situation. Its main limitations are worth understanding so you know when to reach for a different technique.
The halo artifact is the most recognizable limitation. Every specimen viewed in phase contrast is surrounded by a bright luminous ring — it looks like a glow or aura around the edge of each cell or particle. This happens because some diffracted light from specimen edges passes through the phase ring along with the direct light, receiving the same ¼λ shift instead of being left unaltered. The result is constructive interference at the boundary, creating brightness where the physics ideally shouldn’t. The halo is an inherent property of the technique, not a setup error — you can’t eliminate it by adjusting focus or centering. Apodized phase contrast objectives reduce it by adding absorbing bands around the phase ring, but at the cost of some overall brightness. The halo is something you learn to read around rather than fight.
No resolution improvement: A common misconception is that phase contrast “makes things clearer” in the sense of resolving finer detail. It does not. Phase contrast improves contrast — how visible things are — but it does not exceed the standard diffraction limit of light microscopy, roughly 0.2 micrometers (µm). What changes is that structures near that limit, which were invisible in brightfield, become detectable because they now have visible contrast. Numerical aperture still governs actual resolving power.
Shade-off in large or thick structures: Large objects with gradually changing refractive index look brighter in the center than at the edges — the opposite of what you might expect. This “shade-off” effect makes it hard to evaluate the interior of large cells or tissue sections accurately.
Matched optics required: You cannot simply drop a phase objective into any microscope and get phase contrast. You need a condenser with the matching annulus turret, and those annuli must be properly centered. On a compound light microscope not built for phase contrast, you’re missing the condenser hardware entirely.
Frits Zernike and the 1953 Nobel Prize
The physics behind phase contrast had been known for decades before anyone figured out how to exploit it in a microscope. Frits Zernike, a Dutch physicist who worked at the University of Groningen, made the critical leap in the early 1930s: he realized that if you spatially separated direct and diffracted light — using a ring aperture in the condenser — you could then selectively manipulate the direct beam with a matched ring in the objective. He published the principle in 1934, and Carl Zeiss commercialized the first phase contrast microscope in the early 1940s.
Biologists immediately recognized the impact. For the first time, researchers could watch living cells divide, migrate, and respond to stimuli in real time — without the fixation and staining that had been destroying or altering the very phenomena they wanted to study. The technique transformed cell biology and microbiology. Zernike received the Nobel Prize in Physics in 1953 specifically for this invention. For more on how this fits into the broader story of optical biology tools, see our history of the microscope.
Today, phase contrast is standard equipment on virtually every research-grade biological microscope. It represents one of the clearest examples of a physicist’s insight fundamentally changing what biologists can observe — among the most consequential contributions in the long history of microscope types.
Frequently Asked Questions
Can I convert my existing compound microscope to phase contrast?
Most standard compound microscopes cannot be converted without replacing hardware. Phase contrast requires a condenser with a rotating annulus turret — not just an iris diaphragm — and objectives that contain a matched internal phase plate. If your microscope has a basic fixed condenser, you cannot simply add an accessory to get phase contrast. Some manufacturers sell aftermarket phase contrast condenser kits compatible with their own microscope lines; check your make and model’s documentation. In many cases, purchasing a dedicated phase contrast microscope is more cost-effective than retrofitting an older instrument.
Do I need to stain my specimens for phase contrast?
No — and that’s the whole point. Phase contrast was invented specifically to make staining unnecessary for transparent, living specimens. It generates contrast from natural differences in refractive index and thickness between cell structures and their surrounding medium. Specimens viewed under phase contrast remain alive, unfixed, and unstained throughout observation.
Can I use a phase contrast objective on a standard brightfield microscope?
A phase contrast objective will physically fit and produce a usable brightfield image on a non-phase microscope, but you’ll get no phase contrast effect without the matching condenser annulus. Phase contrast requires a condenser with an annulus turret whose ring apertures match the phase rings in each objective. Without the condenser side of the system, you’re just using an expensive brightfield objective.
What do Ph1, Ph2, and Ph3 mean on phase contrast objectives?
The Ph number indicates which annulus size the objective is designed to pair with. Ph1 is the smallest annulus ring, matched to lower-magnification objectives (typically 10×). Ph2 is medium, usually for 20× and 40× objectives. Ph3 is the largest, for high-magnification objectives like 100×. Always match the objective’s Ph number to the same Ph setting on the condenser turret. Mismatching Ph numbers is one of the most common causes of poor phase contrast in practice.
Can phase contrast be combined with fluorescence microscopy?
Yes — many research-grade inverted microscopes support both simultaneously. A common workflow is to use phase contrast for routine live-cell monitoring (identifying healthy, dividing cells) and then switch to the fluorescence channel to examine labeled proteins or organelles in those same cells. The two systems use separate optical paths — transmitted light for phase contrast, epi-illumination for fluorescence — so they don’t interfere with each other optically. The main practical concern is phototoxicity: fluorescence illumination can damage live cells and bleach fluorophores, so phase contrast handles the bulk of observation while fluorescence is used selectively.
How is phase contrast different from darkfield microscopy?
Both techniques reveal specimens that are hard to see under brightfield, but they work on completely different principles. Darkfield microscopy uses oblique illumination — light that would normally bypass the objective — so only specimens that scatter light into the objective are visible, appearing bright against a black background. It’s excellent for surface features and outlines but shows little internal structure. Phase contrast uses interference to reveal internal density differences with a gray background. Darkfield tends to be better for seeing the shape and movement of very small particles; phase contrast is better for seeing internal cell anatomy.
What magnification works best for phase contrast?
Phase contrast is effective across a wide magnification range. 10× is useful for scanning a culture flask or getting an overview. 20× is a common working magnification for general cell culture monitoring. 40× gives good detail for individual cells, nuclei, and organelles. 100× (oil immersion, with a matching oil-immersion phase condenser top) is used for bacteria and fine intracellular detail, though the halo artifact becomes more pronounced at higher magnifications. Most phase contrast work in cell biology happens at 20× and 40×.
Conclusion
Phase contrast microscopy solves one of biology’s most frustrating problems — the invisibility of living cells in brightfield — with an elegant optical trick: separate direct and diffracted light, shift the direct beam by ¼ wavelength, dim it to match the diffracted beam’s amplitude, and let destructive interference do the work. The annular condenser diaphragm and the matched objective phase plate are the two components that make it happen. Understanding the mechanism not only explains what you’re seeing (including that halo) but makes alignment and troubleshooting straightforward rather than mysterious. For a deeper dive into the optics, Nikon MicroscopyU’s phase contrast resource and Molecular Expressions at Florida State are excellent references that go into further optical detail.
Have you tried phase contrast on your own microscope? Whether it’s watching bacteria swim for the first time or seeing your cheek cells in a whole new light, the “before and after” of switching from brightfield to phase is one of those microscopy moments that sticks with you. Tell us what you found in the comments below — we’d love to know what specimens you’re looking at.