
A confocal microscope is a type of optical microscope that uses a tiny pinhole aperture to physically block out-of-focus light, producing razor-sharp “optical slices” through a specimen that can be stacked into a full 3D reconstruction. Unlike a compound light microscope, which captures every plane at once in a blurry superimposition, a confocal microscope builds its image point by point — which means every pixel you see in the final image represents genuine, in-focus signal from a single defined depth. This article walks through how that works: the pinhole principle, the step-by-step optical path, the two main system designs, and the real-world applications and trade-offs you need to understand.
How a Confocal Microscope Works: The Pinhole Principle
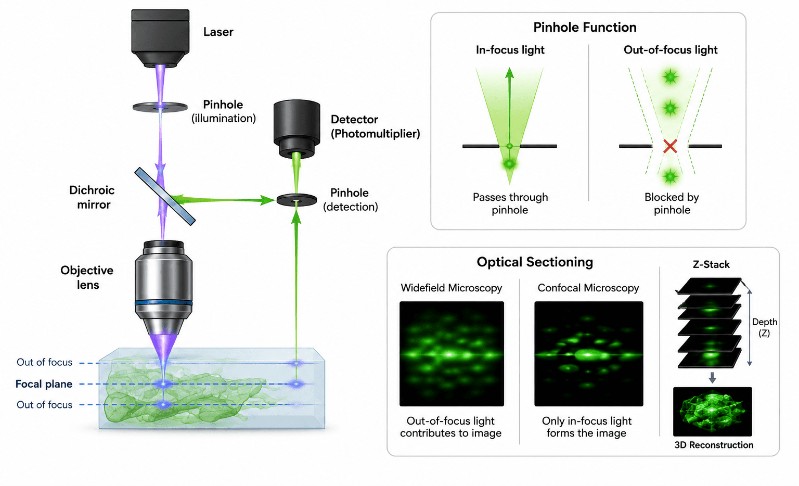
The entire logic of confocal microscopy rests on a single elegant insight: if you block the light that comes from the wrong depth, what’s left is only the light from the depth you care about. The pinhole is how that blocking happens.
What “Confocal” Actually Means
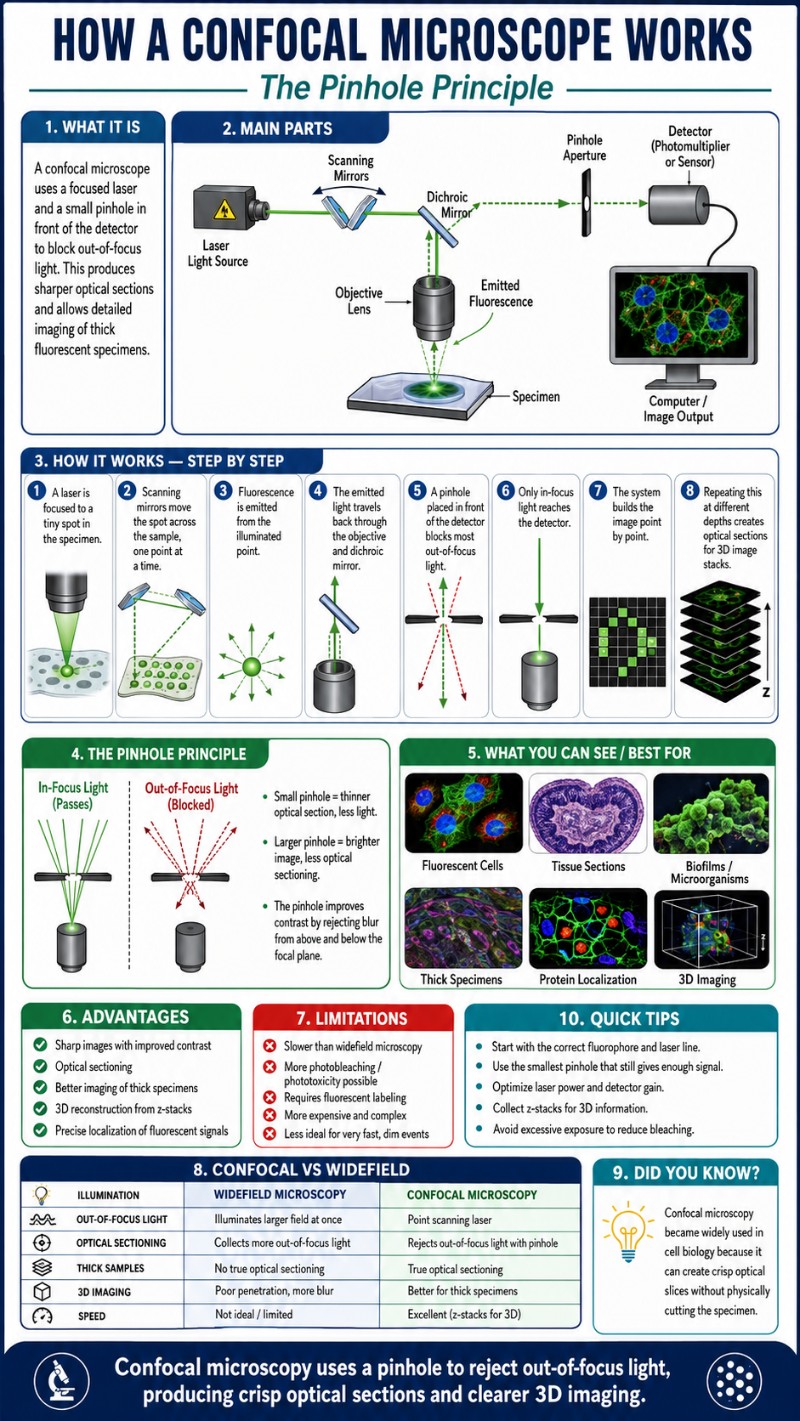
The word “confocal” means conjugate focal planes. In a confocal microscope, the illumination point on the specimen, the focal point inside the sample, and the pinhole aperture in front of the detector are all optically conjugate to each other — they share the same focal position in the optical system. This geometric relationship is what makes the pinhole effective: only light that originates from the precise focal point travels a path that passes through the pinhole. Light from above or below that plane arrives at the pinhole at an angle or spread, so it strikes the edges of the pinhole and is physically stopped.
This is not software processing or digital filtering. It is a mechanical aperture that discards unwanted photons before they reach the detector. The result is a fundamentally cleaner signal.
Why Blocking Out-of-Focus Light Matters
Anyone who has tried to image a thick fluorescent sample on a widefield fluorescence microscope knows the frustration: the image looks like a glowing fog. Fluorescent structures above and below the focal plane all emit light simultaneously, and it all lands on the camera sensor at once, drowning fine detail in a haze of background blur. Switching to a confocal collapses that fog. The same sample, under the same objective, suddenly snaps into crisp isolated structures against a near-black background — the out-of-focus regions go genuinely dark, not just soft. That contrast jump is the first thing everyone notices, and it’s the direct result of the pinhole doing its job.
This also enables optical sectioning: the ability to image a thin plane inside a thick sample without physically slicing it. Combined with the depth of field control that confocal provides, you can non-destructively examine internal structures in a way no conventional widefield instrument can match.
The Optical Path, Step by Step
Here is how light travels through a standard point-scanning laser scanning confocal microscope (LSCM), in order:

- Laser light source. A monochromatic laser beam is used to excite fluorophores in the sample. Common wavelengths are 405 nm (violet/blue), 488 nm (blue), 561 nm (green-yellow), and 633 nm (red), each matched to different fluorescent labels.
- Dichroic mirror (beam splitter). The excitation laser strikes a dichroic mirror — a wavelength-selective beam splitter — that reflects the short-wavelength excitation light toward the specimen but transmits the longer-wavelength fluorescent emission back toward the detector.
- Scanning mirrors (galvanometers). Two small motorized mirrors sweep the focused laser spot across the specimen in a raster pattern — left to right, row by row — building the image one point at a time.
- Objective lens. The objective focuses the laser to a diffraction-limited spot on (or inside) the specimen and collects the fluorescent light emitted back from that exact point. Objective quality and numerical aperture directly determine how tight that focal spot is and how much light is collected.
- Return path through the dichroic. Emitted fluorescence travels back through the objective and passes through the dichroic mirror (it’s now the “right” wavelength to transmit rather than reflect), then arrives at the pinhole.
- Pinhole / confocal aperture. The pinhole — typically set to approximately 1 Airy unit in diameter — allows only the in-focus light through while blocking everything else. This is the heart of the system.
- Detector. A photomultiplier tube (PMT) or, in modern systems, a GaAsP detector or avalanche photodiode, converts the transmitted photons into an electrical signal. The computer records that signal value as one pixel, then the mirrors move to the next point, and the cycle repeats until the full frame is assembled.
The image literally paints itself onto your screen line by line as the mirrors scan — you watch it build rather than seeing it appear all at once. That’s a useful reminder that you’re measuring each point sequentially, not capturing a snapshot.
Confocal vs. Widefield (Regular) Microscope
The core difference between a confocal and a conventional widefield microscope is where the light comes from when the detector records it. In a widefield system — whether a simple bright field microscope or a widefield fluorescence scope — the entire specimen is illuminated at once and the full field is imaged onto a camera simultaneously. Every plane in the sample contributes to every pixel. In a confocal, only one point at a time is illuminated and detected, and the pinhole enforces that only the in-focus signal from that point is recorded.

| Feature | Widefield Microscope | Confocal Microscope |
|---|---|---|
| Illumination | Full field at once | Point by point (or many points — spinning disk) |
| Out-of-focus light | Reaches detector — blurs image | Blocked by pinhole — excluded from signal |
| Optical sectioning | None | Yes — thin Z slices without physical cutting |
| 3D imaging | Not practical for thick samples | Yes — Z-stack reconstruction |
| Speed | Fast (camera frame rate) | Slower (point-scanning) to fast (spinning disk) |
| Cost | Lower | Significantly higher |
| Best for | Thin samples, fast live imaging, routine work | Thick samples, 3D reconstruction, high contrast |
The confocal’s pinhole earns its cost when you have a thick sample, need sharp 3D data, or require the high contrast that out-of-focus rejection delivers. For a thin smear or a monolayer cell culture, a widefield scope often works just as well at a fraction of the price. For context on where confocal fits among different types of microscopes, it sits between conventional light microscopy and super-resolution techniques — more powerful than widefield, still diffraction-limited (see also Britannica’s overview of confocal microscopy for a technical reference).
How Confocal Builds a 3D Image: Optical Sections and Z-Stacks
A single confocal scan captures one optical section — a thin, in-focus plane at a specific depth inside the specimen. Because the pinhole rejects light from every other depth, this slice is sharp even though the tissue above and below it is intact and still present.
To build a 3D image, the microscope moves the focal plane incrementally along the Z-axis (up and down through the sample) and captures a new optical section at each step. This series of sections is called a Z-stack. Software then processes the stack — stacking the sections, aligning them, and rendering them — to produce a 3D reconstruction that you can rotate and explore.
Watching a Z-stack play back is one of the most striking things in modern microscopy. Slice after slice scrolls past, and you experience the sensation of flying straight down through a cell — organelles appearing and disappearing as you move through different depths, exactly like an optical CT scan of a single biological structure. The sections look like genuine cross-sections, not blurry approximations, because each one is clean signal from only that plane.
This is where understanding microscope resolution matters: confocal achieves roughly 180–250 nm lateral (XY) resolution and about 500–700 nm axial (Z) resolution at visible wavelengths. The Z-resolution improvement over widefield is the bigger win — that’s what makes the optical sections possible. Lateral resolution improves only modestly (about 1.4× over widefield) because the confocal is still diffraction-limited. To truly break the ~200 nm diffraction barrier, you need super-resolution techniques such as STED, PALM, or STORM — which are a separate class of instrument beyond confocal.
Point-Scanning vs. Spinning Disk Confocal
There are two mainstream confocal architectures, and the choice between them usually comes down to whether you need maximum resolution and flexibility or maximum speed and gentleness for live cells.
Point-Scanning (Laser Scanning Confocal — LSCM)
The design described above: a single laser spot scanned by galvanometer mirrors, one pinhole, one PMT detector. This is the standard, most flexible design. The pinhole size is adjustable, the optical sectioning is excellent, and a wide range of laser lines and detection channels can be accommodated. The trade-off is speed — raster scanning a full field point by point takes time, typically one to several seconds per frame at high resolution. That’s fine for fixed, stained samples, but inadequate for fast live-cell events. It also delivers a high dose of laser energy to each point during the dwell time, accelerating photobleaching.
Spinning Disk Confocal (Nipkow Disk)
Instead of a single point, a spinning disk (often the Yokogawa design, with paired microlens and pinhole disks) puts thousands of pinholes on a rotating disk that sweeps rapidly across the field. This illuminates hundreds of points simultaneously, and a scientific camera (CCD or sCMOS) captures the full field at once. The result: acquisition speeds of 10–100+ frames per second, and dramatically less laser exposure per point — which means far less photobleaching and phototoxicity. Spinning disk is the go-to for live-cell imaging of dynamic processes. The trade-off: slightly less flexibility in pinhole size, modestly lower out-of-focus rejection compared to the best point-scanning systems, and less straightforward multi-channel detection.
| Feature | Point-Scanning (LSCM) | Spinning Disk |
|---|---|---|
| Speed | Slow (1–5 sec/frame typical) | Fast (10–100+ fps) |
| Photobleaching | Higher (intense dwell per point) | Lower (brief exposure per point) |
| Optical section quality | Excellent | Very good (slightly less rejection) |
| Live-cell suitability | Limited (fixed or slow processes) | Excellent |
| Pinhole flexibility | Adjustable (1–several Airy units) | Fixed disk geometry |
| Relative cost | High | High (but camera-based) |
What Confocal Microscopes Are Used For
Confocal microscopy has become a standard tool across multiple scientific and industrial fields.
Cell biology is the most common application: localizing labeled proteins within organelles, studying how molecules colocalize (or don’t), and imaging thick tissue sections where widefield fails. Immunofluorescence studies — marking specific proteins with fluorescent antibodies and then mapping where they live inside a cell — routinely rely on confocal’s optical sectioning to place structures accurately in 3D.
Neuroscience uses confocal to image neurons, dendritic spines, synaptic connections, and entire brain-slice circuits. The ability to image into thick tissue without physical sectioning is critical when preserving 3D neural architecture for connectivity mapping.
Materials science and industrial inspection use a different confocal mode: reflectance confocal microscopy, which requires no fluorescent dyes at all. Instead of detecting fluorescence, it detects reflected laser light. This maps surface topography with sub-micrometer precision — roughness, step heights, defect geometry on semiconductor wafers, coatings, and optical components. The pinhole principle still provides the Z-sectioning; fluorescence is just one of several possible contrast modes.
Clinical and medical imaging has developed reflectance confocal microscopy (RCM) as a non-invasive technique for examining skin in vivo. Researchers and clinicians use RCM to image skin microstructure — including cellular detail relevant to skin cancer evaluation — without a biopsy, as documented in peer-reviewed dermatology literature via sources such as PubMed/NCBI. These are clinical research and screening tools; they do not replace histopathological diagnosis.
Limitations and Trade-offs
Confocal microscopy is powerful, but it has genuine constraints that matter in practice.
Photobleaching is the most immediate problem. Fluorescent dyes have a finite number of excitation-emission cycles before they are irreversibly destroyed. The focused, intense laser in a point-scanning confocal bleaches dyes rapidly — sometimes visibly within a few scans. Best practice: work fast, use a “throwaway” area of the sample to dial in your settings, then move to the region you actually care about. Lower laser power and compensate with detector gain rather than cranking up the laser.
Phototoxicity is the live-cell corollary: laser energy that bleaches dyes can also damage living cells, generating reactive oxygen species and interfering with the biology you’re trying to observe. Spinning disk systems reduce this significantly, but it’s never zero.
Slow acquisition speed (for point-scanning) is a real operational limit. A large Z-stack at high resolution can take several minutes. During that time the sample can drift, fluorescence can bleach unevenly, and living cells can move. Resonant scanners and spinning disk systems address this at a cost.
Limited penetration depth: light scattering in biological tissue degrades confocal image quality beyond roughly 100–200 µm. Deep-tissue imaging requires two-photon (multiphoton) microscopy, which uses near-infrared excitation to scatter less and reach 500+ µm — a different instrument class entirely.
Cost is a significant barrier. A fully equipped point-scanning LSCM system with multiple laser lines runs from roughly $200,000 to over $500,000, plus maintenance contracts, making it a shared-facility instrument at most institutions.
One point worth being clear about: confocal is not super-resolution. It modestly improves lateral resolution (by roughly 1.4×) and significantly improves axial contrast, but it stays firmly within the diffraction limit. For understanding the distinction between magnification and resolution, confocal improves contrast and Z-sectioning — not raw resolving power beyond what diffraction allows. Also, a wide pinhole setting collapses the optical sectioning advantage: opening the pinhole to 3–4 Airy units to squeeze out more signal essentially converts the confocal into a widefield system. Keep the pinhole near 1 Airy unit to preserve sectioning.
Another common misconception: confocal microscopy is not synonymous with fluorescence microscopy. Most confocal systems use fluorescence, but the defining feature is the pinhole and optical sectioning principle. Nikon’s MicroscopyU confocal introduction covers the distinction well. Reflectance confocal uses no fluorophores at all.

Frequently Asked Questions
What is the difference between confocal and fluorescence microscopy?
They’re not the same thing, though they’re often used together. Fluorescence microscopy describes any technique that detects fluorescent emission from a sample — including widefield epifluorescence scopes. Confocal microscopy describes a specific optical design using a pinhole to reject out-of-focus light. Most confocal microscopes are fluorescence-based, but confocal refers to the pinhole/sectioning principle, not fluorescence itself. Reflectance confocal microscopy uses reflected light with no fluorescent labels at all.
Why is a confocal microscope so expensive?
Several components add up: high-stability laser sources across multiple wavelengths, precision galvanometer scanning mirrors, high-sensitivity photomultiplier or GaAsP detectors, software for 3D rendering and instrument control, and the mechanically and optically precise frame that aligns them all. A fully equipped point-scanning system with three or four laser lines typically costs $200,000–$500,000+ before installation and service contracts. Most users access confocals through institutional core facilities rather than purchasing one outright.
Can a confocal microscope image live cells?
Yes, but with caveats. Point-scanning confocals are slow and deliver concentrated laser energy per point, causing photobleaching and phototoxicity — problems for delicate live cells or fast processes. Spinning disk confocals are far better suited to live-cell imaging: they acquire full frames in milliseconds, distribute laser energy across many points simultaneously, and are much gentler. For very fast or light-sensitive live-cell work, spinning disk (or two-photon for deep tissue) is the standard choice.
Who invented the confocal microscope?
Marvin Minsky, then a graduate student at MIT, invented the confocal microscope and filed his patent in 1957 (granted 1961 as US Patent 3,013,467). His design addressed exactly the out-of-focus light problem described in this article. Practical laser-scanning confocal systems only became widely available in the 1980s, once lasers, fast scanning mirrors, and desktop computers were capable enough to make them usable in real labs.
What’s the difference between confocal and dark field microscopy?
They solve different problems. Dark field microscopy improves contrast for unstained, transparent specimens by excluding direct transmitted light and only detecting scattered light — it works on thin samples without Z-sectioning. Confocal improves contrast in thick fluorescent samples by rejecting out-of-focus light with a pinhole and enables true 3D optical sectioning. The two techniques serve different specimen types and imaging goals.
Does opening the pinhole wider give a better image?
It gives a brighter image, but not a better one — it trades optical sectioning for signal. As you widen the pinhole beyond 1 Airy unit, more out-of-focus light passes through, the Z-sections get thicker and fuzzier, and at very large settings you’ve essentially recreated a widefield fluorescence microscope. If you need more signal, the right solution is to increase detector gain modestly, average multiple frames, or use a brighter/better-matched fluorescent label — not to sacrifice the pinhole’s core advantage.
How deep can a confocal microscope image into tissue?
Typically 100–200 µm into biological tissue before scattering degrades the signal too much to be useful. Highly scattering tissue (such as brain or muscle) limits useful depth even further. For imaging deeper — 500 µm to millimeters — two-photon (multiphoton) microscopy uses near-infrared wavelengths that scatter far less in tissue. Confocal is excellent for imaging cell monolayers, thin sections, and superficial tissue layers; it’s not the right tool for deep whole-organ imaging.
Conclusion
The confocal microscope works by doing something deceptively simple: throwing away the bad light before it reaches the detector. That single pinhole aperture — placed at a plane optically conjugate to the focal point — is responsible for everything that distinguishes confocal from conventional microscopy: the sharp optical sections, the 3D Z-stacks, the high contrast against a black background, and the ability to image inside thick samples without a scalpel. Understanding the pinhole is understanding confocal. The two main variants — point-scanning for precision and spinning disk for speed — are both built on the same principle, optimized for different experimental demands. The trade-offs are real (cost, photobleaching, depth limits, scan speed), but for the problems confocal solves, no other light microscopy technique comes close.
Have you had a chance to use a confocal microscope, or are you trying to figure out whether it’s the right tool for your work? Drop your question or experience in the comments below — we’d love to hear what you’re imaging and what challenges you’ve run into.